Ehlers-Danlos-syndroom
Synoniemen

EDS, Ehlers-Danlos-Meekeren-syndroom, Van Meekeren-syndroom, Fibrodysplasia elastica generalisata, Dermatolyse, Cutis hyperelastica, "rubberen huid", enz.
Frans: Laxité articulaire congénitale multiple
Engels: Danlos-syndroom, Meekeren-Ehlers-Danlos-syndroom, Chernogubov-syndroom, Sack-syndroom, Sack-Barabas-syndroom, Van Meekeren-syndroom I.
Russisch: Chernogubov-syndroom
Definitie / introductie
De Ehlers-Danlos-syndroom (EDS) vat een groep heterogene, genetische Bindweefselaandoeningen samen, veroorzaakt door verstoringen in de synthese van collageen, een structureel eiwit van de Bindweefsel, voorwaardelijke en karakteristieke symptomen van de huid, Gewrichten en interne organen exposeren.
frequentie
De Ehlers-Danlos-syndroom is zeldzaam. De prevalentie in de totale bevolking is 1: 5000; 90% van hen behoort tot hen Typen I, II en III getroffen (30% elk), en ongeveer 10% van type IV De andere vormen worden zelden waargenomen.
De Typen I-III worden autosomale dominantie geërfd, d.w.z. er moet gewoon een defect gen zijn om de ziekte te laten uitbreken. De andere jongens zullen autosomaal recessiefd.w.z. er moeten twee defecte genen zijn, of X-gebondend.w.z. Overdracht van geslachtschromosomen, geërfd.
geschiedenis
dit werd voor het eerst beschreven Ehlers-Danlos-syndroom in jaar 1668 van Job Janszoon uit Meekeren (1611-1666), een chirurg uit Amsterdam. Hij had het symptoom van abnormale overrekbaarheid ontdekt bij een Spanjaard die zijn kinhuid tot aan zijn ogen en over zijn borst kon trekken. Hij nam echter geen andere afwijkingen waar.
Eerste 1891 creëerde de dermatoloog Tsjernogubov een volledige beschrijving van het klinische beeld inclusief de gewrichts- en vasculaire betrokkenheid, daarom in Russische medische
Technische literatuur tot op de dag van vandaag de naam "Chernogubov-syndroom" komt veel voor.
Verdere beschrijvingen volgden 1901 door de Deense dermatoloog Eduard Ehlers (1863-1937) en 1908 door de dermatoloog in Parijs Henri A. Danlos (1844-1912). Het duurde tot 1933 "Ehlers-Danlos-syndroom" zoals de naam van de ziekte heerst.
1949 er waren eerste inzichten in de familiale frequentie van de ziekte en 1972 was een genetische fout die daarmee verband hield Ehlers-Danlos-syndroom ontdekt. In 1986 werd een voorlopige indeling in 10 typen vastgesteld, die in 1997 werd gewijzigd in een vereenvoudigde versie met de onderverdeling in zes hoofdtypen.
oorzaken
De oorzaak van de ziekte is a Genetisch defect. Er is een verandering (mutatie) in de genen waaruit het structurele eiwit bestaat Collageen beschrijven op het menselijk genoom, het DNA. De mutatie leidt tot een veranderde structuur en / of een verminderde synthese van het collageen, wat leidt tot een verminderde sterkte van het gehele bindweefsel.
Beide Typen I en II het is een mutatie in het gen collageen V, waarin Typen IV een mutatie in Collageen III.
Symptomen
Door de verstoorde en verminderde collageensynthese kan de Ehlers-Danlos-syndroom de delen van het lichaam die bijzonder rijk zijn aan bindweefsel: de huid, Gewrichten en Aderen. Omdat het bindweefsel niet sterk genoeg is, is het overrekbaar en scheurt het zeer snel, wat vooral geldt voor te kleine bloedvaten, maar soms ook massale bloeding kan leiden. Een belangrijke complicatie is de vorming van uitstulpingen in de bloedvaten, zogenaamde. Aneurysma's, met het risico van scheuren.
De Belangrijkste symptoom van de huid is het meest uitgesproken Hyperelastische cutisaan de zijkant van de nek, over Gewrichten en ook in gezicht kan tot 4 cm of meer worden opgetild. Na het loslaten springt het onmiddellijk terug naar zijn uitgangspositie, vandaar dat het de naam kreeg "Rubberen huid" dragen. Over het algemeen is de huid merkbaar dun (zoals sigarettenpapier), zacht en fluweelzacht ("Marshmallow-huid").
Wonden vertonen een vertraagde wondgenezing, zodat hechtingen 3 tot 4 keer langer nodig hebben om te genezen. Atrofische of hypertrofische, inferieure ontwikkelen zich vaak uit de naden litteken. Bovendien is er de vorming van met vloeistof gevulde (sappige) huiduitstulpingen (mollusoïde pseudotumoren) op zwaar belaste delen van het lichaam, zoals Knie- en ellebooggewricht, tot de vorming van enkelpads ("Knokkelbeschermers") Aan Achterkant van hand en voet en van knobbeltjes op de hiel-.
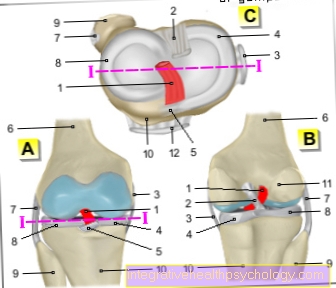
De Gewrichten kunnen overbelast raken (Hyperflexibiliteit), vaak in ongewenste richtingen beweegbaar en zonder sterkte door losgeraakte gewrichtsbanden (Ligament laksheid). Dit kan ongebruikelijke bewegingen veroorzaken, zoals je zou verwachten "Slangenmensen" weet. De gewrichten hebben de neiging Verdraaiingen (Dislocaties) en verkeerde posities. Die worden vooral getroffen schouder- en Enkelgewrichten, de Knieschijf (knieschijf), de Temporomandibulair gewricht (Temporo-mandibulair gewricht) en meer zelden dat Ellebooggewricht. De documentatie van overmobiliteit van de gewrichten (hypermobiliteit) wordt uitgevoerd door de Beightons scoredie hypermobiliteit bevestigt op 5 van de 9 mogelijke punten.
Andere symptomen van de gewrichten zijn algemene gewrichtsproblemen, chronische nekpijn, Actie- en Heuppijn, Gezamenlijke en Spierpijndie moeilijk te behandelen zijn. Soms pijnpunten ("Tender ponits"), die worden gedefinieerd als een gebied dat pijnlijk reageert op drukbelastingen van 4 kg of minder. Bovendien is er een verhoogd risico op fracturen door verminderde botmassa in combinatie met een abnormale botstructuur.
Vanwege de kwetsbaarheid van het bindweefsel van de bloedvaten is er een uitgesproken neiging tot Hematomenspontaan of als gevolg van een trauma,
vooral in gebieden met risico op letsel. Dit wordt gevolgd door een typisch in de getroffen gebieden bruine pigmentatie.
Na blessures merkt men een neiging tot langdurig bloeden met normale coagulatiewaarden. De kwetsbaarheid van grotere bloedvaten kan worden veroorzaakt door inspanning, ongevallen, zwangerschap of geboorte leiden tot ernstige, levensbedreigende bloedingen.
Omdat andere bindweefselstructuren ook inferieur zijn, kan het ook worden Ingewanden (Hernia / inguinale hernia)), kromming van de wervelkolom (Scoliose), Barsten (breuk) van de Darmen en de baarmoeder (Baarmoeder), zakjes (Aneurysma) van bloedvaten en vernauwing van de longen door vrije lucht in de borst (Pneumothorax) komen.
In zeldzame gevallen worden oogveranderingen geassocieerd met EDS, zoals Astigmatisme (astigmatisme) of groene ster (glaucoom) observeren.
diagnose
De diagnose wordt gesteld op basis van het klinische uiterlijk, de symptomen en wordt aangevuld met een familieonderzoek (familiegeschiedenis). Bovendien een Huidbiopsie waarbij het verwijderde huidweefsel wordt onderzocht met een elektronenmicroscoop en de collageenstructuur wordt beoordeeld. De differentiatie in de verschillende soorten Ehlers-Danlos-syndroom vindt plaats door middel van sequentieanalyse van het DNA.
Classificatie / typen
Type I, II: Klassieke kerel; Erfenis: autosomaal dominant; Belangrijkste symptomen: Hyperelasticiteit en kwetsbaarheid van de huid, atrofische littekens, hypermobiliteit van de gewrichten; Oorzaak: stoornis in de vorming van collageen V
Type III: Hypermobiel type; Erfenis: autosomaal dominant; Belangrijkste symptomen: gegeneraliseerde hypermobiliteit van de gewrichten, huidbetrokkenheid (hyperelasticiteit en / of zachte, kwetsbare huid); Oorzaak: stoornis in de vorming van collageen V
Lees meer over dit belangrijke type op: Ehlers-Danlos-syndroom type III
Type IV: Vasculair type; Erfenis: autosomaal dominant; Belangrijkste symptomen: dunne doorschijnende huid, scheuren van de slagaders, darmen en baarmoeder, uitgesproken neiging tot hematoom; Oorzaak: stoornis in de vorming van collageen III
Type V: komt overeen met type I.
Type VI: Kyfoscoliotisch type; Erfenis: autosomaal recessief; Belangrijkste symptomen: verminderde spanning van de Spierstelsel al bij de geboorte ("Floppy baby"), vertraagde ontwikkeling van het vasthouden en ondersteunen van reflexen, laterale buiging van de Wervelkolom (Scoliose), Oorzaak: Lsysl-hydroxylase-deficiëntie
Type VII A / B: Arthrochalastisch type; Erfenis: autosomaal dominant; Belangrijkste symptomen: ernstige gegeneraliseerde hypermobiliteit van de gewrichten met herhaalde dislocaties, congenitale bilaterale heupdislocatie; Oorzaak: Type I collageenstoornis
Type VII C: Dermatosparactisch type; Erfenis: autosomaal dominant; Belangrijkste symptomen: uitgesproken kwetsbaarheid van de huid, slappe huid, Oorzaak: Tekort aan N-terminale procollageen I-peptidase
Therapie en profylaxe
Geen een van de twee oorzakelijk nog steeds een symptomatische therapie momenteel mogelijk is, dus de profylaxe van gevolgschade staat op de voorgrond. Verwondingen en grotere belasting van de gewrichten moeten worden vermeden. Dus moet zeker sport-die gepaard gaan met een verhoogd risico op letsel, worden niet uitgeoefend. Vanwege het verhoogde risico op complicaties zwangerschap en geboorte beide Typen I, II, IV en VI nauwlettend toezicht is vereist.
Het moet ook worden gebruikt bij verkoudheid hoestonderdrukkende therapie en in het algemeen naar de Regulatie van de consistentie van de ontlasting gerespecteerd worden, er is zoiets Colon breuk (Colonbreuk) en een Pneumothorax Kan vermeden worden. Door vroege fysiotherapie, vooral bij kinderen, kunnen de overstrekbare gewrichten worden gestabiliseerd, wat leidt tot verlichting van de symptomen van het gehele bewegingsapparaat.
Wonden Wees extra voorzichtig en operaties mogen alleen in noodgevallen worden uitgevoerd, omdat wondgenezing 3 tot 4 keer langzamer duurt dan normaal.
voorspelling
Geduldig met Ehlers-Danlos-syndroom hebben meestal een normale levensverwachting. De ziekte is echter progressief, dus het leidt tot een steeds toenemende verslechtering van de gezondheid. Huidwonden en gewrichtsdislocaties beïnvloeden de kwaliteit van leven van de patiënt, terwijl het scheuren van de grote bloedvaten levensbedreigend kan zijn.
Levensverwachting
Syndroom van Ehlers-Danlos is een chronische ziekte waarvoor nog geen oorzakelijke behandeling en dus geen genezing bestaat. Dit betekent dat, gezien de huidige stand van de medische technologie, er geen manier is om iets aan de oorzaken van het Ehlers-Danlos-syndroom te doen en deze volledig te genezen. Helaas is men nog steeds niet in staat de optredende symptomen te bestrijden en te behandelen. De betrokken patiënt kan alleen worden aangemoedigd om in het dagelijks leven altijd op te letten, de gewrichten niet te zwaar te belasten en zo mogelijk huidletsel te vermijden. Chirurgische ingrepen mogen alleen worden uitgevoerd in noodgevallen en als er geen geschikte alternatieven zijn.
In de meeste gevallen is het progressief met een toenemende verergering van symptomen en beperkingen in het dagelijkse leven van de patiënt. Afhankelijk van het type ziekte heeft de ziekte verschillende effecten op het leven van de getroffenen. De veranderingen in de gewrichten leiden soms tot artrose en artritis in de vroege kinderjaren, zodat de kinderen later leren lopen en hun voeten vervormd kunnen raken. Met het verhoogde risico op netvliesloslating of retinale bloeding, wordt ook het gezichtsvermogen aangetast.
Hoe sterk de symptomen uiteindelijk bij elk individu worden uitgesproken, hangt sterk af van het type Ehlers-Danlos-syndroom en ze kunnen ook sterk variëren binnen de individuele subtypes. Voor de meeste soorten Ehlers-Danlos-syndroom is de levensverwachting normaal. Bij patiënten met het Ehlers-Danlos-syndroom type IV, dat de bloedvaten aantast, neemt de levensverwachting aanzienlijk af als gevolg van ernstige complicaties, zoals het risico op spontane ruptuur van een slagader, vooral van de hoofdslagader (med. Aortadrank) of de dikke darm.
Het is ongeveer 37 jaar voor vrouwen en 34 jaar voor mannen.
Ook bij het Ehlers-Danlos-syndroom type VI kan een verminderde levensverwachting worden aangenomen.