Taaislijmziekte
Synoniemen in bredere zin
Cystic fibrosis, longen
Engels: mucoviscidose, cystische fibrose
Definitie van cystische fibrose
Cystic fibrosis is een erfelijke ziekte. De overerving wordt medisch autosomaal recessief genoemd. Cystic fibrosis (cystic fibrosis) wordt niet overgeërfd op de geslachtschromosomen X en Y, maar op het autosomale chromosoom 7.
Lees ons algemene artikel over stofwisselingsstoornissen: Stofwisselingsstoornissen - wat betekent het?
De mutatie zit op het zogenaamde CFTR-gen. Recessief betekende dat er twee defecte kopieën van het gen aanwezig moesten zijn om de ziekte te laten uitbreken. Als een persoon een gezonde en gemuteerde genlocatie heeft op het overeenkomstige chromosoom 7, treedt de ziekte niet op.
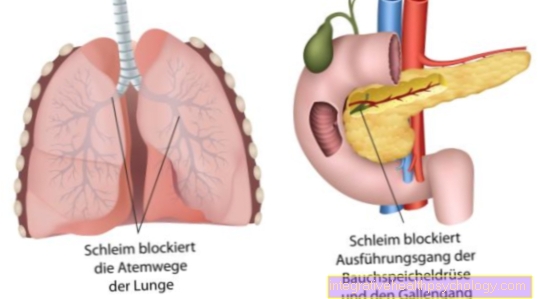
Het resultaat is een pathologisch genproduct. Het daarbij gecodeerde Chloridekanalen zijn kapot. De defecte chloridekanalen leiden tot de vorming van dik slijm in alle exocriene klieren.
Deze exocriene klieren, d.w.z. klieren die hun secretie naar buiten afgeven, zijn onder meer:
- de alvleesklier
- de dunne darm
- het luchtwegsysteem met longen en bronchiën
- de galwegen en
- ook de Zweetklieren
Overzicht
Taaislijmziekte is een Erfelijke ziekte. Het wordt op een zodanige manier geërfd dat het geslacht-onafhankelijk is en alleen met twee defecte genen treedt op. Het is de meest voorkomende autosomaal recessieve overerving.
De gevolgen zijn taaie slijmformaties van alle exocriene klieren, zoals de klieren van de longen, de alvleesklier en ook de zweetklieren. Daar zijn ze op gebaseerd verstoord transport van chloride tussen de binnenkant en de buitenkant van de cel (lees verder: Chloride in het bloed). Het gemuteerde gen is ingeschakeld Chromosoom 7 en veroorzaakt een grote verscheidenheid aan orgaanbetrokkenheid met de overeenkomstige effecten op ademhaling, spijsvertering en voortplanting.
Helaas kan therapie alleen de symptomen verlichten, maar geen genezing bewerkstelligen. De Levensverwachting bij patiënten met cystische fibrose relatief lage.
Omdat het een recessieve erfelijke ziekte is, zijn er mensen die het veranderde gen dragen, maar zelf niet aan de ziekte lijden. Zulke personen worden geroepen Functiedrager of Geleiders, d.w.z. vervoerders. Deze mensen hebben geen cystische fibrose omdat de andere kopie van het gen intact is en de zieke niet sterk genoeg is om te zegevieren.
Ze kan deze defecte kopie van het gen echter wel doorgeven aan haar nakomelingen. Als een gemodificeerd gen al voldoende zou zijn om een ziekte te veroorzaken, zou het een zogenaamde dominante overerving zijn. Zo'n erfenis is bijvoorbeeld te vinden in Chorea Huntington. U kunt meer over deze ziekte lezen in ons onderwerp Chorea Huntington.
Om ongeveer 1:2500 ligt de Ziektecijfer bij pasgeborenen in Duitsland. Vervoerder gaat over iedereen 25. in de Duitse bevolking.
oorzaak

Cystic fibrosis wordt veroorzaakt door een mutatie van een gen op chromosoom 7. Dit chromosoom is een autosomaal chromosoom, geen geslachtschromosoom.
Iedereen heeft 44 autosomale chromosomen (twee identieke versies van elk) en twee geslachtschromosomen. Deze mutatie op chromosoom 7 leidt tot de vorming van defecte chloridekanalen. De reabsorptie (heropname) van chloride uit de klierafscheidingen is niet mogelijk omdat de receptor, het koppelingspunt voor chloride, niet in de klierkanalen is ingebouwd.
In plaats daarvan wordt het gebruikt voor mijnbouw vanwege het onjuiste uiterlijk en de onjuiste structuur. De natuurlijke uitwisseling van chloride via bepaalde chloridekanalen wordt verstoord. Deze zogenaamde kanalen zijn opgebouwd uit eiwitten. In ons DNA is een grote verscheidenheid aan eiwitten gecodeerd. Vanwege het genetische defect van de chloridekanalen is er een gedehydrateerde en taaie productie van slijm uit alle klieren, die hun secretie naar buiten afgeven. Het slijm blokkeert dan gedeeltelijk de kanalen of de luchtwegen in de longen.
Lees ook hierover Chromosoommutatie
Diagnose van cystische fibrose

De typische symptomen die op jonge leeftijd beginnen, zijn baanbrekend bij de diagnose van cystische fibrose.
Dit vermoeden wordt versterkt door een positieve familiegeschiedenis (ziekte van de vader / moeder of naaste familieleden). Een positieve familiegeschiedenis betekent dat er binnen de familie gevallen van cystische fibrose zijn of zijn geweest - aan de kant van de moeder of de vader.
Het ontbreken van pancreasenzymen kan ook in de ontlasting worden gedetecteerd. Eventuele blokkades in de luchtwegen kunnen worden opgespoord door röntgenfoto's van de borstkas te maken.
Een zweettest, die het chloridegehalte van zweet meet, helpt ook bij de diagnose van cystische fibrose. Als een bepaalde waarde wordt overschreden en de andere symptomen ook gelden, ligt de diagnose relatief vast. Vaak merken de ouders zelf het verhoogde zoutgehalte in het zweet van het kind.
Ook het ongeboren kind kan op deze erfelijke ziekte worden getest. Met behulp van een vruchtwaterpunctie (Vruchtwaterpunctie) foetale cellen worden verwijderd en onderzocht op het gemuteerde gen.
Lees meer over het onderwerp: Röntgenonderzoek van het kind
Therapie van cystische fibrose
Iedereen die lijdt aan cystische fibrose, krijgt in één keer advies Cystic fibrosis - polikliniek of advies van Menselijke geneticus (Specialist in erfelijke ziekten) aanbevolen. Deze kunnen helpen om de kwaliteit van leven te verhogen of, als u kinderen wilt hebben, de kans op een ziek kind te berekenen. Mits de ouders vruchtbaar en vruchtbaar zijn.
Anders is de behandeling symptomatisch, omdat de oorzaak, het defecte gen, niet kan worden weggenomen.
Ongeneeslijke ziekte
Taaislijmziekte (taaislijmziekte) is vandaag de dag nog steeds een ongeneeslijke ziekte.
In het geval van cystische fibrose is het belangrijk om voldoende keukenzout (Natriumchloride, NaCl). Mucolyse is uiteraard gericht op. Mucolyse is het oplossen van slijm, vooral in de longen, om het ademen te vergemakkelijken.
Medicijnen en inademing kunnen de symptomen verlichten. Als de longfunctie merkbaar verslechtert, kan zuurstof worden gegeven.
Door intensieve fysiotherapie (fysiotherapie), bijvoorbeeld tikmassage en ademhalingsoefeningen, worden ook de longveranderingen veroorzaakt door cystische fibrose behandeld.
Vaak eindigt de ziekte met een vereiste longtransplantatie. De wachtlijsten zijn echter lang.
Orale toediening van pancreasenzymen en in vet oplosbare vitamines maakt ook deel uit van de therapie. De taak van de alvleesklier moet daarom worden ondersteund, of liever vervangen. In vet oplosbare vitamines zijn A, D, E en K. Ze moeten rechtstreeks in het bloed worden gegeven, omdat ze niet uit voedsel kunnen worden opgenomen vanwege een gebrek aan spijsverteringsenzymen.
Het dieet moet ook veel calorieën bevatten, aangezien slechts een fractie daarvan uit voedsel kan worden verkregen.
Om extra risicofactoren voor complicaties zoals griep of longontsteking te voorkomen, moet het kind worden gevaccineerd. De volgende vaccinaties worden aanbevolen:
- mazelen
- Pneumokokken
- griep
Lees meer over het onderwerp: Superinfectie
Uiteraard vereisen deze maatregelen overleg met een arts, met wie de risico's besproken dienen te worden.
Tegenwoordig wordt er grote hoop op cystische fibrose-therapie gelegd in genetisch onderzoek. Er wordt geprobeerd de ontbrekende genetische informatie in het menselijk genoom te introduceren. We zijn op zoek naar vectoren die deze taak aankunnen. Vectoren kunnen bijvoorbeeld bacterieel of viraal DNA zijn dat erin slaagt de gezonde frequentie in onze genetische samenstelling op te nemen.
De therapeutische aanpak bij ongeboren patiënten wordt momenteel getest. Bij muizen zijn de muizenembryo's er al in geslaagd om het gezonde gen, dat de juiste gensequentie bevatte, door vruchtwaterpunctie (vruchtwaterinoculatie) te introduceren. In deze muizen werd dus het gezonde CFTR-gen geproduceerd. Vruchtwaterpunctie is een punctie en verwijdering van kindercellen uit het vruchtwater via de buikwand van de moeder.
In Duitsland is deze vorm van intra-uteriene (= in de baarmoeder = in de baarmoeder) "therapie" echter verboden.
profylaxe
EEN preventieve maatregel in die zin bestaat het niet omdat het een erfelijke ziekte is.
Er kan echter wel een centrum voor erfelijkheidsadvies bij mensen worden bezocht (meestal te vinden in universitaire ziekenhuizen). Hier wordt berekend hoe groot het risico is om de ziekte door te geven aan kinderen.
Dit advies is altijd nuttig als er een familiegeschiedenis van cystische fibrose is.
Ook een Prenatale diagnostiek is het streven waard. Hier voor de geboorte (d.w.z. prenataal) a Vruchtwateronderzoek (Vruchtwaterpunctie) voerde uit. Foetale cellen (cellen van het kind) worden uit het vruchtwater gehaald en het DNA wordt onderzocht op het gemuteerde gen.
Prognose van cystische fibrose
Helaas is de gemiddelde levensverwachting bij patiënten met cystische fibrose slechts 32-37 jaar. Tegenwoordig wordt de levensverwachting van pasgeborenen die met deze aandoening worden geboren, geschat op ongeveer 45-50 jaar.
De prognose hangt sterk af van de therapie en of deze wordt nageleefd.
De patiënt zelf en zijn motivatie spelen dus een belangrijke rol.